
What is cystic fibrosis?
Cystic Fibrosis is a progressive genetic disease that is inherited in a recessive manner.
Cystic fibrosis is characterized by persistent and frequent lung infections, and limits the ability to breathe over time.
The disease is caused by mutations in the Cystic Fibrosis Transmembrane Conductance Regulator or CFTR gene.
About 75% of new dignosis of cystic fibrosis happens in children below the age of 2 - Source: Healthline
More about the CFTR gene
This gene encodes a protein that is inserted into the cell membranes of the inner lining surface of organs and forms a channel that regulates the movement of salt and water in and out of cells.
Such disease-causing mutations affect the body's ability to efficiently secrete sweat, tears, digestive juices and mucus.
Important: The Cystic Fibrosis report is only applicable to 23andMe v5 chip raw data holders
What happens during cystic fibrosis?

When the CFTR gene functions normally, it aids in the transport of chloride to the cell surface and helps attract water to the surface as well.
This maintains the fluidity of the mucus in most organs.
A mutation in the CFTR leads to improper gene function because of which there is no chloride and water at the cell surface.
The mucus becomes thick and sticky in most organs.
In the lungs, this type of mucus traps bacteria and other microbes eventually leading to lung infections.
Understanding your results
Cystic fibrosis, is an autosomal recessive genetic disease.
This means the disease will only manifest in individuals who receive two mutated copies of the CFTR gene (one from each parent) will get the disease.
More than 500 known mutations of the CFTR gene have been documented to cause cystic fibrosis, although most of them are rare.
How To Analyze Your APOE Gene With Your DNA Raw Data?
What can you do if you have the cystic fibrosis variants?
Though cystic fibrosis can be diagnosed using genetic testing, there are other ways to detect this disease.
These include newborn screening, a sweat test, and clinical evaluation.
Since cystic fibrosis runs in families, couples thinking of having children can take a genetic test and evaluate their results by consulting a physician or genetic counselor.
If the genetic test reveals that you are a carrier of the disease variants, it might be useful to share your result with your family members to help them understand their risk of developing the disease.
Consulting a genetic counselor is always helpful in such cases.
Is cystic fibrosis population specific?
Though there are about 500 mutations known for the genes, the genetic tests done for cystic fibrosis help detect only the most common ones.
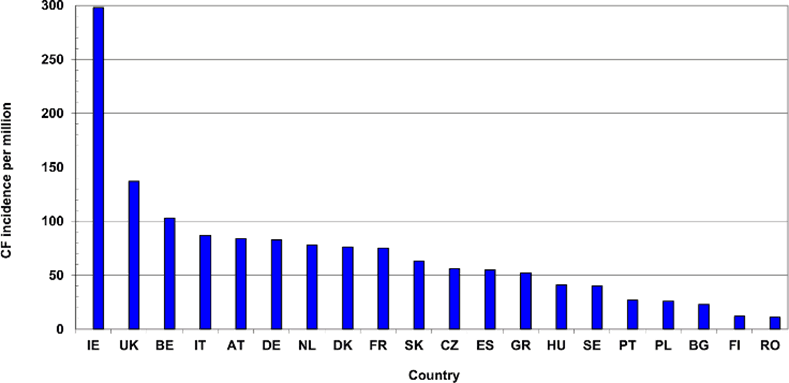
Source: Research Gate
There are more than 70,000 people living with cystic fibrosis in the world today.
In African-Americans and Hispanic-Americans, the carrier rates are 1 in 65 and 1 in 46, respectively.
However, CFTR mutations are much rarer in Asians — with an incidence of 1 in 90.
It is also interesting to note that 1 out of 29 Caucasians, including Ashkenazi Jews, carry one copy of a CFTR mutation.
How is cystic fibrosis inherited?
Since cystic fibrosis is inherited in a recessive manner, an individual must have two copies of the recessive gene, one inherited from each parent.
This means that both parents must have at least one copy of the defective variant.
Individuals with just one copy of the gene are called carriers and do not themselves suffer from the disease.
The effects of having a combination of two different mutations also vary.
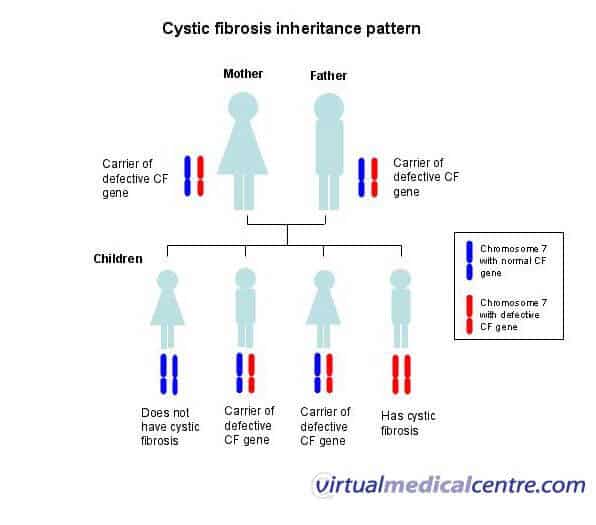
Source: Virtual Medical Centre
Sometimes, a person who has two mutated copies of CFTR might suffer from severe lung and pancreatic disease.
In other cases, lung function may be almost normal but there might be some pancreatic effects.
In some rare cases, there might be only reproductive system effects with little or no noticeable symptoms at all.
Hand-Picked article for you: Have Your 23andMe Raw Data? Use It To Get 500+ Health-Realted Genetic Traits!
How do CFTR mutations affect the reproductive system?
One of the reproductive system effects of CFTR mutation is a congenital bilateral absence of the vas deferens (CBAVD).
Men who have been diagnosed with a low sperm count might be suffering from one CFTR mutation that they should be aware of, as this could be the reason for their infertility if they carry a second, unreported mutation on the other copy of the CFTR gene.
If you are concerned about this, you must consult a health professional.
Which is the most common CFTR gene mutation?
One relatively common CFTR mutation is the deltaF508 mutation.
Individuals with two copies of this mutation have more severe symptoms than those with one copy of this mutation and a copy of another CFTR mutation or two non-deltaF508 mutations.
Can you do a newborn screening for cystic fibrosis?
In many cases, when cystic fibrosis is suspected in newborns, they are screened by assessing the level of a certain enzyme in dried spots of blood.
Other tests are done on babies who test positive for this test in order to rule out or confirm the diagnosis of cystic fibrosis.
In the United States, all newborns are screened for cystic fibrosis as a general rule.
What are the treatment options for cystic fibrosis?
It is important to know which mutation or mutations cause cystic fibrosis in a person as this helps explore the available treatment options.
Today, the treatment of cystic fibrosis focuses primarily on unfixing the mucus, treating infections and supplying adequate nutrition.
In individuals with the G551D mutation, the drug ivacaftor is available for directly treating the dysfunctional CFTR protein.
Currently, the possibility of using ivacaftor for treating CF patients with a handful of other CFTR mutations is being studied. However, it is quite unlikely it would be an effective treatment for the deltaF508 mutation.
Does the 23andMe raw data have the cystic fibrosis mutations?
- DeltaF508 variant
- DeltaI507 variant
- G85E variant
- R334W variant
- R347P/H variant
- A455E variant
- V520F variant
- G542X variant
- S549N variant
- G551D variant
- R553X variant
- R560T variant
- R1162X variant
- W1282X variant
- N1303K variant
- 394delTT* variant
- 621+1G>T variant
- 711+1G>T variant
- 1078delT variant
- 1717-1G>A variant
- 1898+1G>A variant
- 2789+5G>A variant
- 3120+1G>A variant
- 3659delC variant
- 3905insT* variant
- 3849+10kbC>T variant
You can order your Carrier Status Report here
Sources:
- Fast Facts About Cystic Fibrosishealthline.com
- ResearchGate
- https://blog.23andme.com/tag/cystic-fibrosis/
